Estudo traçou a trajetória da epidemia com base em dados genômicos do SARS-CoV-2 obtidos pelo sequenciamento de quase 500 isolados virais de pacientes brasileiros. Resultados foram cruzados com informações sobre viagens aéreas realizadas no período e mortes confirmadas (imagem: divulgação)
Publicado em 05/05/2021
Karina Toledo | Agência FAPESP – Mais de 100 diferentes linhagens do novo coronavírus (SARS-CoV-2) chegaram ao Brasil entre os meses de fevereiro e março de 2020, mas apenas três delas – muito provavelmente vindas da Europa – continuaram a se expandir no país e originaram os mais de 805 mil casos de COVID-19 confirmados até 12 de junho.
Essas três linhagens emergiram nos estados de São Paulo e do Rio de Janeiro entre 22 e 27 de fevereiro e sua transmissão comunitária já estava estabelecida no início de março, bem antes de os órgãos de saúde recomendarem a restrição de viagens aéreas e a adoção de “intervenções não farmacológicas” (NPIs, na sigla em inglês) para conter a disseminação do vírus. O Ministério da Saúde regulamentou em 13 de março os critérios de isolamento social e quarentena, que foram implementados por governadores e prefeitos cerca de uma semana depois. As fronteiras terrestres só foram fechadas em 19 de março e a entrada de estrangeiros por voos internacionais só foi restringida no dia 27 do mesmo mês.
As conclusões são de um estudo apoiado pela FAPESP e divulgado na plataforma medRxiv, ainda sem revisão por pares.
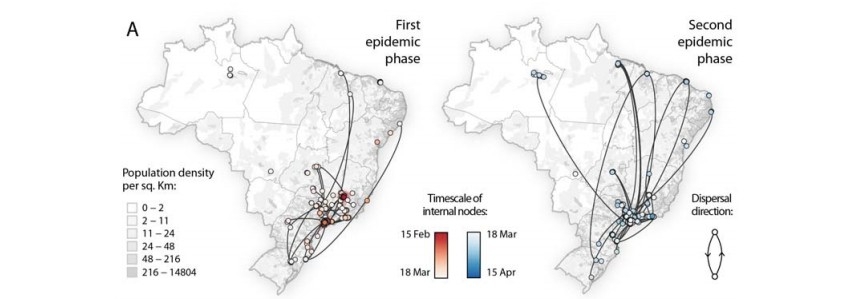
“Nossos resultados evidenciam a existência de duas fases da epidemia no país. A primeira é de transmissão a curta distância, dentro das fronteiras estaduais de São Paulo e Rio. No início de março teve início a fase dois, de longa distância. Ou seja, as pessoas contaminadas nesses dois estados já estavam levando o vírus para as demais regiões do país quando foram adotadas as NPIs”, conta a pesquisadora Ester Sabino, do Instituto de Medicina Tropical da Universidade de São Paulo (IMT-USP), uma das coordenadoras da pesquisa.
Para chegar a essas conclusões, os cientistas usaram um modelo de transmissão orientada pela mobilidade da população. Informações sobre viagens aéreas e sobre as mortes confirmadas por COVID-19 entre fevereiro e abril foram cruzadas com dados genômicos do SARS-CoV-2 obtidos pelo sequenciamento de quase 500 isolados virais de pacientes diagnosticados em 21 dos 27 estados brasileiros (contando o Distrito Federal). O trabalho foi conduzido no âmbito do Centro Brasil-Reino Unido para Descoberta, Diagnóstico, Genômica e Epidemiologia de Arbovírus (CADDE).
Apesar da queda acentuada nas viagens aéreas nacionais após meados de março, os pesquisadores detectaram um aumento de 25% na distância média percorrida por passageiros aéreos no período. Tal fato, segundo os autores, coincidiu com a disseminação do SARS-CoV-2 dos grandes centros urbanos para o resto do país.
“Nossos resultados lançam luz sobre o papel de grandes centros populacionais altamente conectados na ignição rápida e no estabelecimento do SARS-CoV-2 e fornecem evidências de que as atuais intervenções permanecem insuficientes para manter a transmissão do vírus sob controle no Brasil”, afirmam no texto.
O impacto da quarentena
Antes das medidas de isolamento social serem adotadas, a taxa de contágio do SARS-CoV-2 no Brasil estava em torno de 3. Isso significa que cada infectado transmitia o vírus, em média, para três outras pessoas, o que favorecia o crescimento exponencial da doença.
Embora tenham sido implementadas quando a transmissão comunitária já estava estabelecida e o vírus já havia cruzado as fronteiras paulistas e fluminenses, as restrições da quarentena conseguiram – em um primeiro momento – conter significativamente a disseminação da doença.
O modelo de transmissão orientada pela mobilidade mostra que a taxa de contágio chegou a ficar abaixo de 1 nas cidades de São Paulo e Rio de Janeiro logo após a adoção das NPIs, o que evitou o crescimento exponencial do número de casos e o colapso dos hospitais.
Mas, à medida que a adesão da população ao isolamento diminuiu, a taxa de contágio foi lentamente aumentando para valores entre 1 e 1.3 e não mais baixaram. Especialistas em epidemiologia afirmam que somente quando a taxa de contágio se estabiliza abaixo de 1 durante algumas semanas o crescimento no número de casos e de mortes começa a desacelerar.
Por meio de análises de filogeografia – que combinam os dados de sequenciamento do genoma viral com as informações do local em que ocorreu a transmissão – os pesquisadores identificaram que 104 linhagens do SARS-CoV-2 entraram no Brasil, a maioria oriunda dos Estados Unidos.
Do total de genomas sequenciados no Brasil, 75% pertencem a três linhagens ou clados de origem europeia: 186 genomas (38%) correspondem ao “clado1"; 161 (33%) são do “clado 2”; e 19 (4%) se inserem no “clado 3”.
“É possível que as outras linhagens que identificamos não tenham conseguido se expandir porque quando elas entraram no Brasil já haviam sido implementadas as medidas de isolamento social. Mas é bem provável que, à medida que mais isolados virais forem sequenciados no país, clados diferentes sejam identificados. No Reino Unido, onde já foi feito o sequenciamento de mais de 20 mil amostras de pacientes com COVID-19, já foram identificadas mais de mil entradas do novo coronavírus”, conta Sabino.
Como explica a pesquisadora, o genoma do SARS-CoV-2 tem cerca 30 mil pares de bases (que formam as cadeias do RNA viral). Caso o vírus que infecta um indivíduo sofra uma mutação na posição 200 da cadeia de RNA, por exemplo, todas as pessoas que se contaminarem a partir desse paciente vão carregar a mesma marca no genoma viral. “Ao cruzar esses dados com informações sobre a data e o local em que as amostras foram coletadas conseguimos traçar a trajetória da epidemia, que ainda está apenas no começo”, afirma Sabino.
Segundo a pesquisadora, ainda será preciso sequenciar mais amostras da região Norte do país para determinar, por exemplo, a origem da linhagem que se disseminou fortemente por estados como Amazonas e Pará. “O que já sabemos é que os deslocamentos fluviais entre as cidades amazônicas contribuíram muito para espalhar o vírus”, diz.
Na avaliação de Sabino, esse tipo de estudo ajuda a entender como uma epidemia evolui e quais são as principais rotas de transmissão. “Esse conhecimento talvez sirva de lição para que em uma situação futura as medidas sejam tomadas mais precocemente e de forma mais efetiva.”
O artigo Evolution and epidemic spread of SARS-CoV-2 in Brazil pode ser lido em https://www.medrxiv.org/content/10.1101/2020.06.11.20128249v1.
Fonte: https://agencia.fapesp.br/33396
