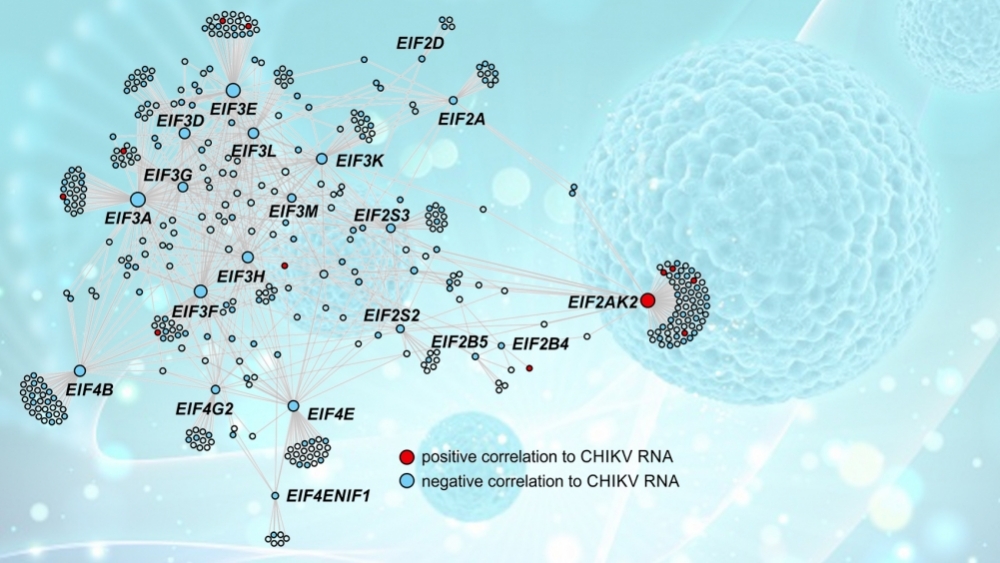
Using a systems biology approach, Brazilian researchers identified several genes that can be explored as therapeutic targets and as biomarkers of predisposition to chronic joint pain (image: PLOS Pathogens; kjpargeter/Freepik)
Published on 05/12/2021
By Karina Toledo | Agência FAPESP – Computational tools applied to biology are revolutionizing the study of what happens inside cells during an infection, helping scientists to understand disease mechanisms and contributing to the identification of potential therapeutic targets.
An example is a study published in PLOS Pathogens describing how Brazilian researchers analyzed blood cells from patients infected with chikungunya virus. With the aid of techniques such as complex network analysis, artificial intelligence and machine learning, the group identified gene signatures associated with the disease – sets of genes whose expression is altered by interaction with the virus. They then investigated the role played in cells by the involved genes and determined the importance of these genes to efforts to combat the virus.
Conducted in Brazil, the research was supported by FAPESP. The principal investigator was Helder Nakaya, a professor at the University of São Paulo’s School of Pharmaceutical Sciences (FCF-USP). Researchers at the same university’s Biomedical Science Institute (ICB-USP) and its Ribeirão Preto Medical School (FMRP-USP), as well as colleagues at Butantan Institute and the Public Health Central Laboratory of Sergipe, among others, also contributed.
“We also identified a set of genes that show during the acute phase whether the patient is likely to develop chronic arthralgia [joint pain and inflammation], a relatively common condition in people infected with chikungunya. However, this finding has yet to be confirmed by future research based on a larger number of samples,” Nakaya told Agência FAPESP.
The article describes the results of analyses using blood samples from 39 subjects born in Sergipe, a state in Northeast Brazil, and infected during the 2016 epidemic. These results were compared with data for a control group comprising 20 uninfected subjects from the same region.
The first step was an analysis of the samples’ transcriptomes, comprising all molecules of messenger RNA (which encodes proteins) as well as noncoding RNAs (which do not produce proteins but perform a regulatory role in the genome) expressed in red blood cells, white blood cells and platelets. By quantifying the transcript levels in the samples, the researchers were able to measure the activity levels of 20,000 genes and determine whether their expression increased or decreased during infection; these findings were also compared with results for the control group.
“We focused on protein-coding genes [which express messenger RNA] because their role is easier to interpret. It’s relatively easy to know whether they encode cell receptors or transcription factors, for example. In this way, we were able to enhance our understanding of the pathogenesis of chikungunya – how the virus affects cells and which defense systems are activated in response to infection,” Nakaya said.
Their analysis revealed the mechanism by which immune cells trigger inflammatory processes to eliminate the virus. The proteins responsible for this immune response are collectively known as the inflammasome, a multiprotein intracellular complex that can be formed by different proteins and result in the production of different proinflammatory molecules. The researchers found that the mediating factor in chikungunya virus infection is the enzyme caspase-1.
The finding was validated in experiments with mice performed in partnership with Dario Zamboni, Full Professor at FMRP-USP. Both Nakaya and Zamboni are affiliated with the Center for Research on Inflammatory Diseases (CRID), one of the Research, Innovation and Dissemination Centers (RIDCs) funded by FAPESP. The CRID is hosted by FMRP-USP.
They found that chikungunya virus infection in mice genetically modified to not express caspase-1 did not lead to the release of a proinflammatory molecule called interleukin-1 beta (IL-1β), whereas it did in wild-type (non-GM) mice.
Similar but different
Having identified the gene signatures of chikungunya virus infection, which involve thousands of genes whose expression is altered by the disease, the group compared the results with those obtained using samples from patients infected with dengue virus.
“We noticed that the gene signatures of both diseases were similar to a considerable extent but that some genes were specific to chikungunya. These can be explored further in drug development research,” Nakaya said.
In another analysis, the researchers compared the gene expression profile of patients infected with chikungunya virus with the profile of patients suffering from rheumatoid arthritis, an autoimmune disease characterized by chronic joint inflammation.
“In this case, our aim was to discover the difference between arthritis caused by the virus and autoimmune arthritis. We wanted to identify any genes that were specific to the viral infection,” Nakaya explained.
Combined analysis of three gene signatures showed that 949 genes were involved in rheumatoid arthritis alone, 632 in dengue alone, and 302 in chikungunya alone. Seven genes were connected with all three conditions: OAS1, C1QB, ANKRD22, IRF7, CXCL10, IFI6, and IFIT3.
The researchers then performed gene coexpression analysis using CEMiTool, a software package developed by Nakaya with FAPESP’s support. The aim of this analysis was to understand how the genes interact with each other within the complex network that exists in every cell, forming signaling pathways and metabolic pathways.
“We were able to identify eight main coexpression modules [genes with similar response profiles]. We also identified the network hubs – the genes with the most connections and, hence, the most promising targets to explore for drug development purposes,” Nakaya said.
The data used in the study, both the raw data and analysis results, are available in a public repository and can be downloaded by anyone free of charge, he stressed, as can the code for the software package mentioned earlier, so that others can reproduce the results.
“Our research enabled us to produce a list of potential therapeutic targets, and we’re now cross-referencing these findings with a database of active compounds. This cross-referencing is being done computationally but on the basis of experimental data compiled from published studies that have pointed to drugs capable of interfering with these genes of interest,” Nakaya said.
The group will also continue to analyze the transcripts found in the samples from 39 patients infected with chikungunya virus but are now focusing on noncoding RNAs.
Nakaya was supported by FAPESP via a Regular Research Grant and a Young Investigator Grant. The study was also supported via CRID and another RIDC, the Center on Toxins, Immune Response and Cell Signaling (CeTICS).
The article “Systems analysis of subjects acutely infected with the Chikungunya virus” by Alessandra Soares-Schanoski et al. can be read at the following address: https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1007880.
Source: https://agencia.fapesp.br/31470
