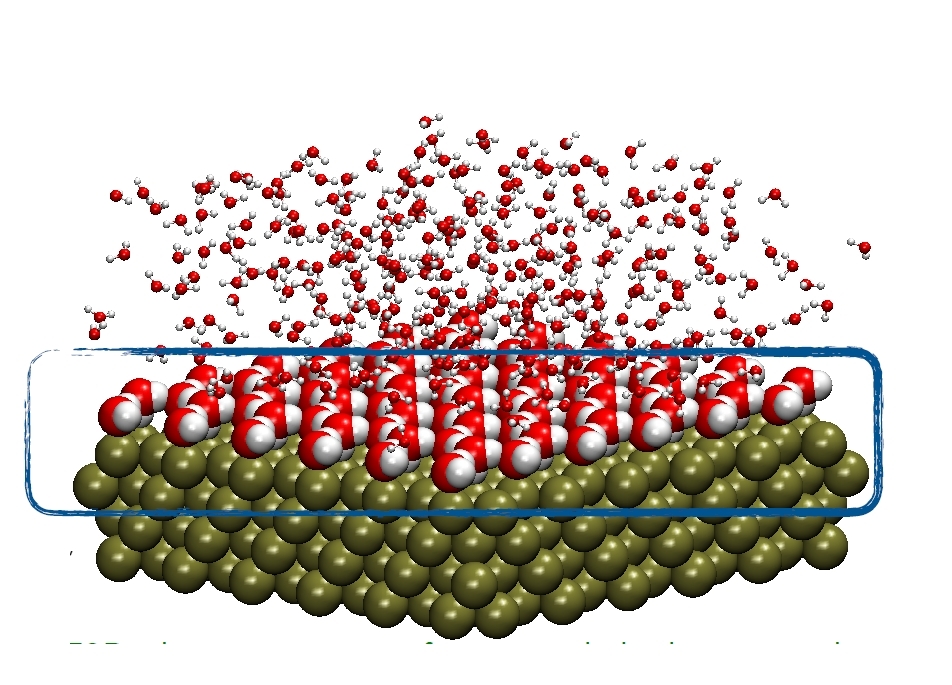
Upgrading fuel cells is one of the possible technological applications of a theoretical study led by the young Brazilian researcher Luana Sucupira Pedroza (image: atomistic simulation of the interface between a metal [green spheres] and water [red and white spheres]/ Luana Sucupira Pedroza)
Published on 05/13/2021
By José Tadeu Arantes | Agência FAPESP – The need to produce cleaner energy in the context of the global climate crisis is the motivation for an innovative research project presented at the Frontiers of Science Symposium organized by FAPESP and the Max Planck Society (MPG) in November 2018 in São Paulo, Brazil.
The study, conducted by Luana Sucupira Pedroza, a professor at the Federal University of the ABC (UFABC) in Brazil, aims to optimize electrochemical reactions in fuel cells and other devices. These reactions, whose main output of interest is hydrogen, involve solid-liquid interactions. The focus of the research project is the use of computer simulation to understand what happens at the interface between the two systems.
The study is supported by FAPESP via its Young Investigator Grant Program and an agreement with Germany’s Max Planck Society.
“Although electrochemistry has been known for a long time, we still don’t fully understand how the solid-liquid interface behaves on the microscopic scale or what we can do to improve the efficiency of the reaction. The goal of our research is basically to understand what happens at the interface between a solid and a liquid, particularly water,” Pedroza told Agência FAPESP.
In addition to its contribution to the development of fundamental science, explaining what happens in the experimental context, the study has obvious potential for technological application in that it can help engineers choose the best solid materials or design new interface configurations.
“We work in the theoretical field with computer simulation, but we’re closely connected to experimental research. We collaborate with experimental physicists in Germany, and among other things, our simulations set out to explain the results they obtain but don’t always understand. Atomistic simulations are indispensable for this kind of explanation,” Pedroza said.
“Our study involves what are known as ab initio simulations because they’re based on first principles. This means we don’t use experimental data to design our models. We use our knowledge of fundamental physical processes and ab initio calculation. Of course, approximations are always necessary, but the goal is to describe real phenomena with a minimum of assumptions so as not to confine the interpretation to the established empirical repertoire. We want to be able to predict new possibilities.”
The phenomena clearly have to do with the solid material used. Pedroza has mainly studied palladium, platinum and gold, metals of interest both as electrodes and as catalysts. However, the behavior of the interface depends on geometry as well as the type of material. “Interaction with water changes depending on how the material is cut, or in other words depending on the cleavage plane. The behavior of the water does not depend only on the metal used. It also depends on the geometry of the metallic surface. This is a very important point that simulations can explain. The configuration of the water molecules at the interface is determined by the surface geometry,” she said.
Water molecules (H2O) are coplanar: the single oxygen and dual hydrogen atom centers are in the same plane, and in this plane, the centers are positioned in a V shape with the oxygen at the vertex and the hydrogens at the two tips, forming an angle of approximately 104.5 degrees. Depending on the metal and the geometry of its surface, i.e. the coordinates of the cleavage plane, the molecules in the first water layer can be parallel to the surface or skewed so that the hydrogens are closer to or farther from the surface. These different configurations strongly influence the reaction.
Because its atoms form a V shape, water is an electric dipole. Several properties of water, including the fact that it is an excellent solvent, derive from its dipolarity. In this regard, it differs from carbon dioxide (CO2), for example. In CO2, the three atoms are aligned linearly, with the carbon in the center and one oxygen on either side, preventing polarization.

“Water’s properties also depend on its local organization, on how each molecule relates to neighboring molecules,” Pedroza said. “This behavior, in turn, depends on the charge-charge relationships, a classical component and purely electrostatic, and on the other hand on the relationships between the atoms’ electronic orbitals, which determine a quantum component. In ab initio simulations, the atomic nuclei are treated as classical particles, while the electronic orbitals are treated as quantum systems. This has been the standard procedure since the late 1980s. However, because hydrogen is a very light atom, the procedure doesn’t always constitute a good approximation of the real phenomenon. This is where our study innovated, by including quantum effects for the nuclei as well. This is the key aspect of our collaboration with Germany. Our German partner is a specialist in this.”
Making everything quantum mechanical in a model comprising several hundred molecules, in which metal-water and water-water interactions coexist and compete, entails using what are known as “Feynman path integrals”, a technique that requires heavyweight computational processing and substantial machine time. FAPESP provided Pedroza with funding for this via its Young Investigator Grant Program. To increase the processing capacity available from computer clusters at UFABC and the National Scientific Computation Laboratory (LNCC) in Petrópolis, Rio de Janeiro State, which she already uses in terms of machine hours with time limits, Pedroza is in the process of buying a cluster destined to prioritize her research project. This new cluster will run on the UFABC premises.
In addition to quantum effects for electrons and nuclei, the model will also include the effects of temperature and external voltage applied to the metal. This part of the model requires highly complex simulations, which will offer the scientific community highly realistic descriptions of electrochemical processes. “Assembling all these variables is something that’s never been done before. We expect to do it for the first time,” Pedroza said.
Source: https://agencia.fapesp.br/29491
